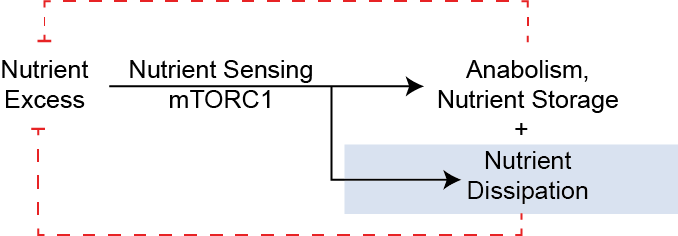
The obesity pandemic is a leading cause of ill health, and is a risk factor for many chronic diseases including diabetes, liver disease and cardiovascular disease. While most approved anti-obesity therapies focus on reducing energy intake, the other way to promote negative energy balance is to elevate energy expenditure and nutrient dissipation. But how is nutritional status sensed? In one framing, the center of nutrient sensing is the protein kinase complex mTORC1. While mTORC1 is widely appreciated to mediate nutrient storage, I think of mTORC1 as responding to nutrient and energy excess by engaging both anabolic and catabolic pathways.
First lets consider the counterfactual, that mTORC1 promotes primarily anabolic pathways, what might we expect from that framing:
- Increased triglyceride storage in tissues with activated mTORC1. This could include adipose and liver tissues.
- Nutrient absorption might be increased, for example mTORC1 could promote gastrointestinal transport to more efficiently absorb nutrients when activated.
- Overall increased nutrient storage in models with activated mTORC1, and reduced in conditions of reduced mTORC1 activity. This could be due to positive energy balance (e.g. by reduced energy expenditure and/or increased appetite).
As I describe below, none of this occurs.
Physiological processes governed by mTORC1: A Dual-Flux Model of Anabolism and Catabolism
The anabolic roles of mTORC1 are very well established (See 1 for a review). These include promoting:
- Protein synthesis: particularly protein translation via 4EBP phosphorylation, S6K-dependent eIF4B phosphorylation, and eEF2-kinase phosphorylation and activation. mTORC1 also enhances ribosome biogenesis via both promoting rRNA transcription and translation of ribosomal mRNAs. mTORC1 also enhances m^6^A‑dependent post‑transcriptional control. This selectively increases the translation and stability of specific (usually growth‑promoting) mRNAs 2.
- Nucleotide synthesis: promoting the synthesis of purine and pyrimidine biosynthesis both via activation of the pentose phosphate pathway and promoting folate flux via ATF4-dependent transcriptional activation. There is also evidence of mTORC1–S6K–CAD–mediated activation of de novo pyrimidine synthesis and mTORC1‑dependent regulation of SAM synthesis (the major methyl donor required for nucleotide synthesis) via Myc-dependent transcriptional activation of MAT2A 3.
- Lipid synthesis: activating SREBP1c to promote de novo lipogenesis and triglyceride synthesis. A recent paper also shows that cholesterol biosynthesis is promoted via USP20 dependent stabilization of HMGCR, the rate limiting enzyme of cholesterol synthesis 17.
- Glycogen synthesis: mTORC1 activation causes induction of the protein phosphatase 1 targetting subunits PTG4 and PPP1R3B5. The resultant dephosphorylation and activation of glycogen synthase (and inactivation of glycogen phosphorylase) promotes glycogen synthesis in vitro.
- Preventing autophagy: The catabolism of proteins via autophagy is also strongly attenuated by mTORC1 activity, in part by phosphorylation of ULK1 and Atg13 as well as TFEB inhibition, causing transcriptional downregulation of autophagic genes. In this way mTORC1 functions to prevent protein catabolism.
What these anabolic processes have in common is that they are all dependent on the presence of available nutrients (amino acids, folates, fatty acids) as well as are all generally energetically costly. Notably in all of these circumstances, these nutrients are effectively consumed or stored. Let's now review some of the key mTORC1-dependent nutrient consuming processes.
The dissipation of nutrients is driven by mTORC1
Promoting anabolic function alone would be predicted to cause nutrient dissipation, as amino acids, fatty acids, glucose and micronutrients are stored as macromolecules. However, in addition to that anabolic depletion, there is considerable evidence that, paradoxically, these nutrients are also catabolized in states of mTORC1 activation.
Amino acid oxidation is positively regulated by mTORC1
The two most relevant steps for amino acid catabolism are glutamate catabolism into $\alpha$-ketoglutarate (several amino acids are catabolised via transaminases into glutamate and a carbon skeleton) and the resulting ammonia detoxification by the urea cycle. Glutamine is a store of amino groups that can then be used for non-essential amino acid biosynthesis. Two publications report the activation of glutaminolysis and glutamate oxidation in mTORC1-activated cells 6 7, though the later has since been retracted. mTORC1 inhibition by rapamycin also causes reduced activity of BCKDH, the rate limiting step of BCAA catabolism 8 9. Finally knockout of the GATOR1 component Nprl2 )(a negative regulator of mTORC1 signaling) in muscle also resulted in increased expression of BCAA catabolism genes 10.
Glycolysis is promoted via mTORC1-dependent HIF1 activation
Transcriptional profiling of TSC knockout MEFs showed upregulation of most glycolytic genes, along with increased glucose uptake and lactate efflux 11. Tracers showed that this included mTORC1-dependent flux of glucose through both glycolysis the oxidative PPP pathway. One proposed mechanism is that mTORC1 activates HIF1$\alpha$, normally a sensor of hypoxia to promote glycolysis. Similarly deletion of the inhibitory GATOR1 compelx (Nprl2 knockout in muscle) also resulted in increased anaerobic glycolysis 10. Cao et al. investigated glycolytic rates in kidneys and kidney epithelial cells and showed that mTORC1 inhibition reduced lactate production, while proximal tubule-specific Tsc1 knockout increased lactate production, suggesting a positive role for mTORC1 in anaerobic glycolysis 12. Together these data support a model that mTORC1 activation shifts glucose catabolism towards anaerobic glycolysis in multiple tissues, in line with the cataplerotic oxidation of amino acids.
mTORC1 promotes skeletal muscle fuel oxidation.
In muscle tissue, mTORC1 is activated by obesity @Khamzina2005b @Tremblay2007, concordant with a switch towards oxidative muscle fibers @Lexell1995a @Goodpaster2001 @Dube2008. Cell and animal models of mTORC1 activation have been developed via deletion of its negative regulators TSC1 and TSC2. These studies show that mTORC1 activation is sufficient for mitochondrial biogenesis, ATP production and whole-body energy expenditure @Cunningham2007 @Ramanathan2009 @Koyanagi2011 @Bentzinger2013 @Guridi2015.
Ketone body disposal is enhanced in mTORC1-activated muscle tissues
Ketogenesis is an alternative catabolic pathway for excessive fatty acids. In our model, we would predict that under chronic mTORC1 activation in liver we would observe increased ketone body production and that would be dissipated in ketolytic tissues. Indeed we do observe more rapid ketone body clearance in muscle Tsc1 knockout mice 13. Where things are less straightforward is in the ketogenesis step, where there are conflicting data whether liver Tsc1 knockout does 14 15 or does not 16 suppresses fasting ketone body levels.
mTORC1 as an Engine of Diet-Induced Thermogenesis
Increased energy expenditure is a normal physiologic mechanism to compensate for elevated caloric intake.
Clinical studies have shown that energy expenditure can adjust to under- or overfeeding @Acheson1988 @Leibel1995a. This is consistent with the observation that obese individuals have higher energy expenditure than lean subjects @James1978 @Ravussin1982 @Ravussin1986 @Leibel1995a @Delany2013. This process of diet-induced thermogenesis (DIT) is an adaptation to higher macronutrient levels and functions to limit weight gain. In a small human study, acute suppression of energy expenditure by induced by fasting was inversely associated with the ability to lose weight on chronic calorie restriction @Meng2020. This suggests that diet-related adaptive thermogenesis may be more prognostic than the association between basal metabolic rate and obesity risk, which is minimal @Luke2009 @Rimbach2022. The mechanisms underlying DIT are largely unclear, but previous studies have shown that impairment of MC4R or β-adrenergic signaling blocks DIT @Rothwell1979 @Butler2001 @Bachman2002 @Berglund2014. We have shown that mTORC1 activity is necessary for diet-induced thermogenesis (DIT) and is sufficient for increased energy expenditure and resistance to diet-induced obesity and metabolic disease.
Thermogenesis in Brown Adipose Tissue
Several papers demonstrate a role for mTORC1 in promoting BAT thermogenesis @Liu2016b @Labbe2016 @Lee2016a @Tran2016 @Magdalon2016,
Why do mouse models of mTORC1 activation typically appear as catabolic?
Intracellular scarsity and suppression of conservatory mechanisms
In this framing, mTORC1 causes a nutrient dissipation flux. In normal cells, a drop in nutrients would inhibit mTORC1, which then acts as a circuit breaker to this flux. However, in constitutive models (e.g., TSC1/2, GATOR1, or Sestrin deletions, Rheb/Rag activation mutants, PRAS40 deletion), the circuit breaker is removed and nutrient dissipation continues. As such, even as internal nutrient concentrations ($[N]$) fall, the cell continues to drive protein and lipid synthesis and nutrient catabolism. This intracellular scarcity is a state of starvation in the midst of plenty; the cell has the machinery to grow, but the raw materials are being dissipated faster than they can be replenished.
The enhanced export of nutrients in models of mTORC1 activation
mTORC1 governs several transport processes in physiology. In contrast to the lens that mTORC1 activity simply promotes anabolism, there are at least three clear examples where mTORC1 promotes both synthesis and export of macronutrients (liver VLDL production, milk macronutrient secretion, and enterocyte lipid uptake), often with the balance being towards prmoting nutrient dissipation.
mTORC1 activation models cause reduced, not enhanced liver fat
Just as mTORC1 promotes the synthesis of cholesterol and lipids via the USP20–HMGCR axis 17, it simultaneously coordinates their export in the form of VLDL. In spite of an anabolic framing of lipid levels in the liver, Tsc1 knockout livers can clear hepatic lipid stores despite high synthetic rates. This paradoxical reduction in liver fat highlights a critical underappreciated role for mTORC1 in lipid export, challenging the notion that mTORC1 primarily drives lipid accumulation.
Liver triglycerides can be thought of a balance between fatty acid uptake (primarily from adipocytes) but also from remnant lipoprotein particles), fatty acid oxidation (and ketone body production/release), and with triglyceride efflux in the form of VLDL particles. Evidence from genetic models underscores mTORC1's essential role in VLDL secretion. Liver-specific deletion of Tsc1 (leading to constitutive mTORC1 activation) results in markedly reduced hepatic triglyceride content, even during fasting, compared to controls 18. Conversely, inhibition of mTORC1 via liver-specific Raptor knockout increases liver TAG accumulation, as secretion is impaired. In vivo secretion assays demonstrate that mTORC1 activation doubles VLDL-TAG export rates, while inhibition halves it, directly linking mTORC1 activity to lipid efflux 18. This cell-autonomous effect persists in isolated hepatocytes, where mTORC1-deficient cells show reduced TAG secretion despite normal synthesis, confirming that export, not just production, is mTORC1-dependent.
The mechanism f or mTORC1 control of VLDL efflux involves mTORC1-driven phosphatidylcholine (PC) synthesis, a key component of VLDL particles. mTORC1 regulates the rate-limiting enzyme phosphocholine cytidylyltransferase α (CCTα), promoting PC synthesis. In Tsc1 knockouts, elevated CCTα activity and PC levels facilitate VLDL assembly and secretion, reducing lipid buildup 18. Restoring PC synthesis in mTORC1-inhibited models (e.g., via CDP-choline supplementation) rescues both secretion defects and hepatosteatosis, demonstrating PC as the critical mediator 18. This dual role underscores the limitations of viewing mTORC1 solely as a promoter of lipid storage; instead, it produces a dissipative flux that prevents excess accumulation.
Lactation and Nutrient Delivery
During lactation, the mammary gland demands high nutrient flux to synthesize and secrete milk components. Prolactin, the master regulator of lactation, activates mTORC1 in mammary epithelial cells to promote milk protein synthesis and epithelial cell proliferation 19. This mTORC1 activation may translating detected nutrient excess into milk production, further supporting the dissipative model where mTORC1 drives anabolic processes in response to nutrient availability.
Beyond mammary epithelial cells, adipocyte mTORC1 also plays a crucial role in mobilizing lipids for milk production. Activation of mTORC1 in adipocytes increases milk lipid content in lactating mice, while reducing the size of mammary adipocytes, demonstrating how mTORC1 signaling enhances nutrient delivery to the mammary gland 20. Similar to the phenotype in the liver, chronic activation of mTORC1 causes less lipid remaining in the mammary gland and increased secretion, in this case into milk.
Intestinal Lipid Absorption and Chylomicron Export
The intestinal enterocyte faces a unique challenge among lipid-handling cells: it must absorb, repackage, and export dietary fat without accumulating toxic levels of free fatty acids or unesterified lipids intracellularly. The enterocyte handles this by re-assembling absorbed lipids into chylomicron particles that are secreted basolaterally into the intestinal lymph. If left unpackaged, absorbed lipids can be transiently stored in cytoplasmic lipid droplets, creating a dynamic partitioning between export and storage that determines the rate of dietary lipid delivery to peripheral tissues. mTORC1 signaling appears to govern this partitioning in favor of export to the tissues, consistent with the dissipative flux model observed in liver and mammary gland.
Direct evidence for mTORC1 control of chylomicron production comes from enterocyte-like Caco-2 cell models. Knockdown of encoding Raptor significantly decreased expression of lipogenic enzymes (FASN, DGAT1, DGAT2), lipoprotein assembly genes (APOB, MTTP), fatty acid uptake machinery (FATP2, DBI), and prechylomicron transport vesicle components (VAMP7, SAR1B), resulting in repressed secretion of apoB-containing triacylglycerol-rich lipoprotein particles 21. Conversely, cells with constitutively active mTORC1 showed enhanced chylomicron-like particle secretion21. These findings demonstrate that mTORC1 promotes the entire chylomicron assembly pipeline from fatty acid activation and TAG re-esterification, through apoB48 lipidation by MTP, to the COPII-like vesicular transport of prechylomicrons from the ER to the Golgi.
As in the liver, the phosphatidylcholine axis is a critical mechanistic link. mTORC1 drives PC synthesis for particle coating, but the stoichiometry of the phospholipid surface coat must be balanced. Excess PC relative to other membrane lipids impairs, rather than enhances, particle formation, suggesting that mTORC1's role is to maintain sufficient flux through the Kennedy pathway to support chylomicron assembly without overwhelming the system.
The net effect of mTORC1 activation in the enterocyte is thus strikingly parallel to the hepatic and mamamary epithelial phenotype: rather than just synthesizing lipid, mTORC1 activation accelerates the transit of fat through the enterocyte and into the lymph. mTORC1 activation in the enterocyte shifts the balance away from cytoplasmic lipid droplet storage and toward chylomicron secretion, all to move dietary lipid rapidly through and out of the absorptive cell.
Effects on Appetite and Energy Intake
If this About Nutrient Excess, and if so which nutrients?
Implications
Leveraging nutrient dissipation for cancer growth
How does this square with mTORC1's effects on immune function
Aging and Chronic Disease
References
-
Ben-Sahra, Issam, and Brendan D. Manning. 2017. “mTORC1 Signaling and the Metabolic Control of Cell Growth.” Current Opinion in Cell Biology 45:72–82. doi:10.1016/j.ceb.2017.02.012. ↩
-
Meyer, Kate D., and Samie R. Jaffrey. 2014. “The Dynamic Epitranscriptome: N6-Methyladenosine and Gene Expression Control.” Nature Reviews Molecular Cell Biology 15(5):313–26. doi:10.1038/nrm3785. ↩
-
Villa, Elodie, Umakant Sahu, Brendan P. O’Hara, Eunus S. Ali, Kathryn A. Helmin, John M. Asara, Peng Gao, Benjamin D. Singer, and Issam Ben-Sahra. 2021. “mTORC1 Stimulates Cell Growth through SAM Synthesis and m6A mRNA-Dependent Control of Protein Synthesis.” Molecular Cell 81(10):2076-2093.e9. doi:10.1016/j.molcel.2021.03.009. ↩
-
Lu, B., Dave Bridges, Y. Yang, K. Fisher, A. Cheng, L. Chang, Z. X. Meng, J. D. Lin, M. Downes, R. T. Yu, C. Liddle, R. M. Evans, and A. R. Saltiel. 2014. “Metabolic Crosstalk: Molecular Links between Glycogen and Lipid Metabolism in Obesity.” Diabetes 63(9). doi:10.2337/db13-1531. ↩
-
Uehara, Kahealani, Won Dong Lee, Megan Stefkovich, Dipsikha Biswas, Dominic Santoleri, Anna Garcia Whitlock, William Quinn, Talia Coopersmith, Kate Townsend Creasy, Daniel J. Rader, Kei Sakamoto, Joshua D. Rabinowitz, and Paul M. Titchenell. 2024. “mTORC1 Controls Murine Postprandial Hepatic Glycogen Synthesis via Ppp1r3b.” The Journal of Clinical Investigation 134(7):e173782. doi:10.1172/JCI173782. ↩
-
Csibi, Alfredo, Gina Lee, Sang Oh Yoon, Haoxuan Tong, Didem Ilter, Ilaria Elia, Sarah Maria Fendt, Thomas M. Roberts, and John Blenis. 2014. “The mTORC1/S6K1 Pathway Regulates Glutamine Metabolism through the Eif4b-Dependent Control of c-Myc Translation.” Current Biology 24(19):2274–80. doi:10.1016/j.cub.2014.08.007. ↩
-
Csibi, Alfred, Sarah-Maria Fendt, Chenggang Li, George Poulogiannis, Andrew Y. Choo, Douglas J. Chapski, Seung Min Jeong, Jamie M. Dempsey, Andrey Parkhitko, Tasha Morrison, Elizabeth Petri Henske, Marcia C. Haigis, Lewis C. Cantley, Gregory Stephanopoulos, Jane Yu, and John Blenis. 2013. “The mTORC1 Pathway Stimulates Glutamine Metabolism and Cell Proliferation by Repressing SIRT4.” Cell 153(4):840–54. doi:10.1016/j.cell.2013.04.023. ↩
-
Zhen, Hongmin, Yasuyuki Kitaura, Yoshihiro Kadota, Takuya Ishikawa, Yusuke Kondo, Minjun Xu, Yukako Morishita, Miki Ota, Tomokazu Ito, and Yoshiharu Shimomura. 2016. “mTORC1 Is Involved in the Regulation of Branched‐chain Amino Acid Catabolism in Mouse Heart.” FEBS Open Bio 6(1):43–49. doi:10.1002/2211-5463.12007. ↩
-
Neishabouri, S. Hallaj, S. M. Hutson, and J. Davoodi. 2015. “Chronic Activation of mTOR Complex 1 by Branched Chain Amino Acids and Organ Hypertrophy.” Amino Acids 47(6):1167–82. doi:10.1007/s00726-015-1944-y. ↩
-
Dutchak, Paul A., Sandi J. Estill-Terpack, Abigail A. Plec, Xiaozheng Zhao, Chendong Yang, Jun Chen, Bookyung Ko, Ralph J. Deberardinis, Yonghao Yu, and Benjamin P. Tu. 2018. “Loss of a Negative Regulator of mTORC1 Induces Aerobic Glycolysis and Altered Fiber Composition in Skeletal Muscle.” Cell Reports 23(7):1907–14. doi:10.1016/j.celrep.2018.04.058. ↩↩
-
Düvel, Katrin, Jessica L. Yecies, Suchithra Menon, Pichai Raman, Alex I. Lipovsky, Amanda L. Souza, Ellen Triantafellow, Qicheng Ma, Regina Gorski, Stephen Cleaver, Matthew G. Vander Heiden, Jeffrey P. MacKeigan, Peter M. Finan, Clary B. Clish, Leon O. Murphy, and Brendan D. Manning. 2010. “Activation of a Metabolic Gene Regulatory Network Downstream of mTOR Complex 1.” Molecular Cell 39(2):171–83. doi:10.1016/j.molcel.2010.06.022. ↩
-
Cao, Hongdi, Jing Luo, Yu Zhang, Xiaoming Mao, Ping Wen, Hao Ding, Jing Xu, Qi Sun, Weichun He, Chunsun Dai, Ke Zen, Yang Zhou, Junwei Yang, and Lei Jiang. 2020. “Tuberous Sclerosis 1 (Tsc1) Mediated mTORC1 Activation Promotes Glycolysis in Tubular Epithelial Cells in Kidney Fibrosis.” Kidney International 98(3):686–98. doi:10.1016/j.kint.2020.03.035. ↩
-
Cousineau, Cody M., Detrick Snyder, JeAnna R. Redd, Sophia Turner, Treyton Carr, and Dave Bridges. 2024. “Reduced Beta-Hydroxybutyrate Disposal after Ketogenic Diet Feeding in Mice.” BioR$\Chi$ivdoi:10.1101/2024.05.16.594369. ↩
-
Sengupta, Shomit, Timothy R. Peterson, Mathieu Laplante, Stephanie Oh, and David M. Sabatini. 2010. “MTORC1 Controls Fasting-Induced Ketogenesis and Its Modulation by Ageing.” Nature 468(7327):1100–1106. doi:10.1038/nature09584. ↩
-
Kucejova, Blanka, Joao Duarte, Santhosh Satapati, Xiaorong Fu, Olga Ilkayeva, Christopher B. Newgard, James Brugarolas, and Shawn C. Burgess. 2016. “Hepatic mTORC1 Opposes Impaired Insulin Action to Control Mitochondrial Metabolism in Obesity.” Cell Metabolism 16(2):508–19. https://www.cell.com/cell-reports/abstract/S2211-1247(16)30725-2. ↩
-
Ebru S., and Michael J. Wolfgang. 2021. “mTORC1 Activation Is Not Sufficient to Suppress Hepatic PPARα Signaling or Ketogenesis.” The Journal of Biological Chemistry 297(1):178–83. doi:10.1016/j.jbc.2021.100884. ↩
-
Lu, Xiao-Yi, Xiong-Jie Shi, Ao Hu, Ju-Qiong Wang, Yi Ding, Wei Jiang, Ming Sun, Xiaolu Zhao, Jie Luo, Wei Qi, and Bao-Liang Song. 2020. “Feeding Induces Cholesterol Biosynthesis via the mTORC1–USP20–HMGCR Axis.” Nature 588(7838):479–84. doi:10.1038/s41586-020-2928-y. ↩↩
-
Quinn WJ III, et al. mTORC1 stimulates phosphatidylcholine synthesis to promote triglyceride secretion. J Clin Invest. 2017;127(11):4126–4136. doi:10.1172/JCI96578 ↩↩↩↩
-
Wu, Z., Q. Li, S. Yang, T. Zheng, J. Shao, W. Guan, F. Chen, and S. Zhang. 2022. “Energy Deprivation-Induced AMPK Activation Inhibits Milk Synthesis by Targeting PrlR and PGC-1α.” Cell Communication and Signaling 20(1):25. doi:10.1186/s12964-022-00830-6. ↩
-
El Habbal, Noura, Allison C. Meyer, Hannah Hafner, JeAnna R. Redd, Zach Carlson, Molly C. Mulcahy, Brigid Gregg, and Dave Bridges. 2021. “Activation of Adipocyte mTORC1 Increases Milk Lipids in a Mouse Model of Lactation.” bioRxiv doi:10.1101/2021.07.01.450596. ↩
-
Kaur H, Moreau R. Chylomicron production is repressed by RPTOR knockdown, R-α-lipoic acid and 4-phenylbutyric acid in human enterocyte-like Caco-2 cells. J Nutr Biochem. 2022;108:109087. doi:10.1016/j.jnutbio.2022.109087 ↩↩
-
Zhang P, Csaki LS, Ronquillo E, Baufeld LJ, Lin JY, Gutierrez A, Dwyer JR, Brindley DN, Fong LG, Tontonoz P, Young SG, Reue K. Lipin 2/3 phosphatidic acid phosphatases maintain phospholipid homeostasis to regulate chylomicron synthesis. J Clin Invest. 2019;129(1):281–295. doi:10.1172/JCI122595. PMCID: PMC6307960 ↩
-
Arif A, Terenzi F, Potdar AA, Jia J, Sacks J, Chen A, Mishra P, Nussinov R, Fox PL. EPRS is a critical mTORC1–S6K1 effector that influences adiposity in mice. Nature. 2017;542(7641):357–361. doi:10.1038/nature21380 ↩